Les propriétés pharmacologiques d’un médicament peuvent aider à prévenir son devenir dans l’organisme et son effet thérapeutique mais en réalité, des mécanismes plus complexes sont mis en jeu.
Le thiopental illustre bien ce propos : en effet, il a une demi-vie d’élimination longue mais sa durée d’action est courte après injection d’un bolus unique. On comprend ici l’origine des modèles pharmacocinétiques, censés être plus proches de la réalité.
Rappels
-
- Pharmacocinétique = étude des relations entre la dose administrée d’un médicament et sa concentration obtenue dans le sang.
"ce que l’organisme fait à la substance"Le devenir d’une substance dans l’organisme dépend de phénomène d’absorption, de distribution et d’élimination.
-
- Pharmacodynamie = relation entre la concentration dans le sang et l’effet obtenu.
"ce que la substance fait à l’organisme"La relation dose / concentration / effet est exprimée sur le schéma ci–dessous et met en lumière les principes de pharmacocinétique et pharmacodynamique :

Devenir d’un médicament injecté dans l’organisme
Après une injection d’un bolus intraveineux d’un médicament, l’évolution de sa concentration dans le plasma va être liée à des phénomènes de distribution et d’élimination.
Sa concentration plasmatique augmente brutalement puis décroit progressivement, au cours du temps, générant un gradient de concentration entre le sang et les tissus. La différence de concentration va donc entrainer un passage du compartiment central jusqu’aux compartiments périphériques jusqu'à l’obtention d’un état d’équilibre des concentrations. Ce phénomène se produit d’abord dans les organes et tissus les mieux vascularisés puis dans les moins bien vascularisés jusqu'à l’obtention d’un milieu d’équilibre à terme.
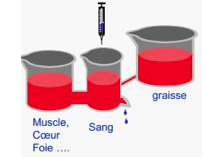
La baisse de la concentration sanguine et donc des concentrations à terme ne s’effectuent plus que grâce à l’élimination de la substance.
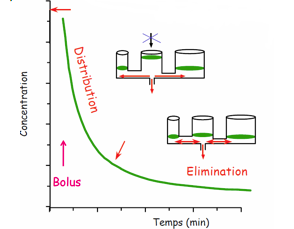
Décroissance de la concentration des agents IV au cours du temps. Au bout d’un moment, la concentration sanguine ne diminue plus sauf si il y a élimination de la substance
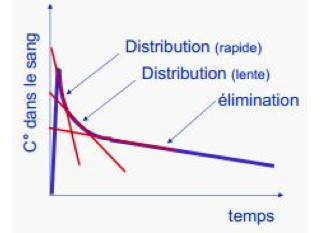
La courbe de décroissance des concentrations plasmatiques peut être décomposée en une somme d’exponentielles qui vont constituer le modèle pharmacocinétique de l’agent.
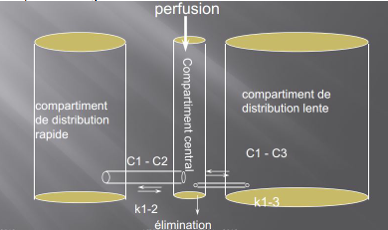
 La cinétique de la plupart des agents anesthésiques obéit à un modèle pharmacocinétique à trois compartiments : un compartiment central (le sang circulant et les tissus richement vascularisés), et deux compartiments périphériques où l’agent se distribue plus ou moins rapidement.
La cinétique de la plupart des agents anesthésiques obéit à un modèle pharmacocinétique à trois compartiments : un compartiment central (le sang circulant et les tissus richement vascularisés), et deux compartiments périphériques où l’agent se distribue plus ou moins rapidement.
L’élimination ne se fait qu’à partir du volume central et il n’y a pas de transferts entre les compartiments périphériques.
La notion de compartiment est relativement artificielle et ne correspond toutefois pas à des organes clairement identifiés.
En résumé :
Le bolus s’équilibre quasi instantanément dans le compartiment central plasmatique, puis sa distribution se fait dans les compartiments périphériques, avant d’être éliminé depuis le compartiment central.
Les médicaments d’anesthésie se répartissent la plupart du temps dans 3 compartiments.
On conçoit ici le trajet des médicaments de l’anesthésie.
Mais pourquoi la durée d’obtention d’un effet (par exemple la perte de consicence) diffère d’une molécule à l’autre ?
Compartiment site d’action et délai d’action
Le transfert entre le sang et le système nerveux central n’est pas immédiat. Il existe un retard de diffusion des produits au niveau du compartiment, lié au passage de la barrière hémato-encéphalique.
Pour illustrer cette réalité clinique, un quatrième compartiment a été créé, appelé le site d’action, site effet ou biophase.
Lorsque le médicament s’équilibre entre le compartiment sanguin et les autres compartiments, notamment celui de la biophase, on peut définir une constante d’équilibration, appelée ke0.
Ke0 relie la concentration plasmatique (Cp) à la concentration au site d’action (Ce).
Les valeurs de la ke0 sont les suivantes :
- keO (min-1) sufentanil : 0,112
- ke0 rémifentanil : 0,595
- ke0 propofol Marsh : 0,26
- ke0 propofol Schnider : 0,456
A une même intensité d’effet peuvent correspondre deux concentrations sanguines différentes : on appelle ça l’hystérésis d’effet.

Représentation d’un modèle pharmacocinétique tri-compartimental.
Ce modèle est paramétré par les volumes de ses compartiments V1, V2, V3, par ses constantes de transfert (k12(exprime la rapidité de diffusion de V1 à V2), k21(de V2 à V1), k13, k31) et ses constantes d’élimination(k10) et par ke0.
Le même principe s’applique lors de l’élimination du médicament hors de l’organisme. Elle se fait à partir du compartiment central et est décrite par la constante ke (e pour élimination) ou k10 (du compartiment 1 vers l’extérieur).
La distribution peut varier en fonction du volume de distribution (plus élevé par exemple chez l’obèse) et l’élimination s’il existe une insuffisance hépatique ou rénale, le foie et le rein étant les principaux organes d’épuration de l’organisme.
De même que les propriétés physico-chimiques de la molécule vont intervenir dans les mouvements vers le site d’action.
Il s’agit de :
- - la liposolubilité : seules les molécules liposolubles peuvent franchir la barrière hémato-encéphalique.
- - la ionisation : la forme non ionisée est habituellement liposoluble et franchit les membranes.
- - la fixation protéique : en effet, seule la forme libre de la molécule, c'est-à-dire non fixée sur les protéines va pouvoir franchir les barrières physiologiques. Ainsi, un patient en hypoalbuminémie, avec un taux de protéines bas, va voir sa forme libre de médicament augmentée et l’effet pharmacologique pour une même dose administrée sera donc plus important.
En resumé :
Le médicament une fois injecté met du temps à passer la barrière hémato-encéphalique et donc à atteindre le cerveau. Ce délai entre l’injection plasmatique et l’effet obtenu au niveau du cerveau est définit par une constante, dite d’équilibration : la ke0. Voila pourquoi lorsqu’on injecte un certain bolus, l’effet n’est pas immédiat.
Les propriétés du médicament en lui-même, l’état général du patient (débit cardiaque, âge, poids…) influent aussi sur la rapidité d’obtention de l’effet, de même que sur l’arrêt de l’effet.
Ainsi, même si la concentration plasmatique diminue, l’effet peut perdurer. A un même effet peuvent donc correspondre plusieurs concentrations plasmatiques. C’est ce qu’on appelle l’hystérésis d’effet.
Mais comment peut-on évaluer la durée du réveil en fonction des paramètres expliqués ci-dessus ?
Durée d'action
La demi-vie contextuelle ou temps de demi-décroissance ou Context-Sensitiv half-time (CSHT) est le temps nécessaire pour que la concentration, à l’arrêt de la perfusion diminue de moitié. Elle correspond à l’exponentielle la plus lente du modèle pharmacocinétique et donc à la décroissance finale de la concentration et en cela, elle n’est pas très intéressante en pratique clinique.
Un paramètre pharmacocinétique plus pertinent a fait son apparition : il s’agit du temps de décroissance. C’est le temps nécessaire à l’arrêt de la perfusion pour que la concentration diminue jusqu’au niveau de la concentration probable de réveil. Les logiciels d’AIVOC le calculent en temps réel et en fonction de la durée de perfusion et des vitesses utilisées.
C’est toutefois à l’anesthésiste de déterminer la concentration de réveil pour chaque patient. Le temps de décroissance varie en fonction de la durée de perfusion, d’autant plus avec les agents qui s’accumulent rapidement.

Temps de décroissance affiché par défaut par le module lors de l’AIVOC Propofol.« la concentration de réveil site effet lors d’une AIVOC propofol est de 1,5μg/ml, elle sera atteinte chez ce patient en 3 minutes ».
En fait, la concentration de réveil correspond à la concentration au site effet relevée lors de la perte de conscience.
Supposons que ce patient ait perdu connaissance à une concentration de 2,4µg/ml au site effet, on peut rentrer cette concentration correspondante comme la concentration de réveil et le module d’AIVOC recalcule automatiquement le temps de décroissance (ici de 2 minutes).


En resumé :
Le paramètre le plus important lors d’une anesthésie en mode AIVOC pour prédire le réveil est le temps de décroissance. Il peut être « prescrit » par l’anesthésiste et est déterminé lors de l’induction, à la perte de conscience. Il suffit de le prendre en note, pour le reprogrammer de façon plus ajustée au patient.
Comment toutefois prévoir les concentrations à programmer dès l’induction pour être au plus proche du patient ?
Les modèles pharmacocinétiques auraient peu d’intérêt dans la pratique de l’AIVOC s’ils en restaient à de simples analyses d’évolution des concentrations.
Ainsi, à quoi correspondent plus exactement les concentrations entrées dans les tableaux d’abaques ?
Comment peut-on être sur qu’elles vont être adaptées au patient ?