Cette étape va se décliner en trois partie : la description de la création d’un modèle pharmacocinétique, des détails concernant les valeurs des paramètres pharmacocinétiques et la description de chacun des modèles pharmacocinétiques… cette étape est la dernière avant le passage à l’application clinique.
Création d’un modèle pharmacocinétique
En pratique, pour déterminer un modèle pharmacocinétique, on regroupe tous les prélèvements sanguins effectués chez différents sujets en fonction de la dose reçue, et on effectue des analyses statistiques dites par régression sur l’ensemble des points (de prélèvements). On recherche alors les paramètres pharmacocinétiques moyens observés chez le plus grand nombre de sujets. Cette méthode est appelée pharmacocinétique de population et permet, entre autre, de définir les facteurs de variations significatifs pour une molécule donnée (l’âge, le poids…).
La performance du système peut ensuite être évaluée pour voir si les concentrations prédites correspondent à celles mesurées. C’est ainsi que certains modèles jugés trop imprécis ne seront jamais intégrés dans des bases permettant d’effectuer de l’AIVOC.
Les valeurs des différents paramètres pharmacocinétiques peuvent être fixes ou dépendantes d’un certain nombre de covariables : poids, âge, masse maigre….
Valeurs des paramètres pharmacocinétiques
A une concentration donnée au site d’action correspond un effet recherché que l’état d’équilibre soit atteint ou pas. La courbe reliant la concentration d’un médicament à cet effet est en général une courbe sigmoïde.
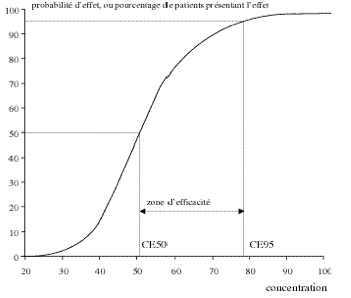
Courbe décrivant la relation concentration/effet d'un agent, dite courbe de Hill.
La zone d'efficacité se situe entre la concentration efficace chez 50 % des patients et la concentration efficace chez 95 % d'entre eux.
On observe ainsi sur cette courbe qu’au bout d’un certain temps, et donc au-delà de certaines concentrations, l’effet est maximal et ne varie plus.
Concrètement, une fois l’effet maximum atteint, on aura beau augmenter les concentrations cibles sur le module d’AIVOC, l’effet ne variera plus.
Pour évaluer l’évolution de la concentration au site effet, deux paramètres sont à prendre en compte :
- - le temps d’accès à l’équilibre de la concentration au site effet
- - le délai d’obtention du pic de concentration = time to peak effect = Tmax
Le temps d’accès à l’équilibre au site effet correspond à 5 à 7 demi-vie d’élimination (= 5 à 7 fois le T1/2 vie ke0). Cette valeur dépend donc de la ke0.
Le délai d’obtention du pic de concentration est le temps écoulé depuis le bolus jusqu’à l’obtention du pic pharmacologique et donc jusqu’à l’obtention de l’effet.
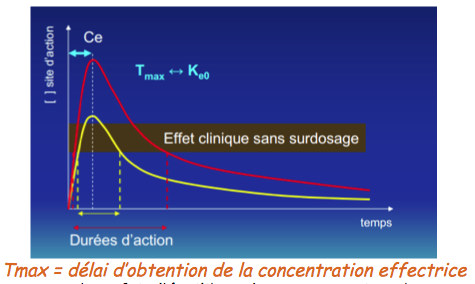
On pourrait penser qu’une fois l’équilibre de concentration obtenu au site effet, le pic de concentration est atteint. Mais il n’en est rien. Pour deux médicaments ayant la même ke0, si la distribution d’un médicament B dans les compartiments périphériques est plus rapide qu’un médicament A, alors le Tpeak sera obtenu plus rapidement.
Le Tpeak est donc un paramètre qui représente plus le comportement pharmacocinétique initial d’un médicament.
En resumé :
Le médicament idéal serait donc celui qui a la ke0 la plus élevée, le Tpeak le plus court et la demi-vie contextuelle la plus courte, le tout avec peu d’effets secondaires.
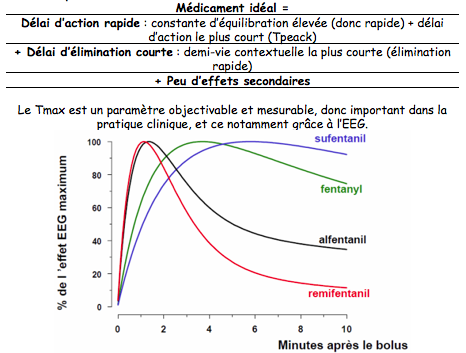
Temps nécessaire à l’obtention de l’effet EEG maximum
La relation entre concentration et effet pharmacodynamique peut-être résumée par la notion de concentration efficace (Ce). Le Tpeak est donc un paramètre qui représente plus le comportement pharmacocinétique initial d’un médicament.
La concentration efficace impose la notion de probabilité générée par des calculs automatisés. L’administration en mode AIVOC permet d’obtenir la concentration souhaitée au site effet (par administration d’un bolus calibré pour remplir le compartiment central) puis de la maintenir le temps voulu (par une perfusion exponentiellement décroissante de façon à compenser la redistribution et l’élimination).
Le modèle pharmacocinétique permet de prédire la concentration plasmatique d’après la dose administrée, le temps écoulé et les paramètres du modèle. L’utilisation à l’envers grâce au logiciel informatique et aux algorithmes contenus va permettre d’administrer une certaine dose sur un temps T pour obtenir la concentration et donc l’effet voulu.
Description des modèles pharmacocinétiques.
Le propofol possède deux modèles pharmacocinétiques et leur utilisation ne s’impose pas dans les mêmes circonstances. Ces deux modèles sont tricompartimental, ont un vaste volume de distribution, une clairance métabolique élevée,.
Le modèle de Marsh
C’est un modèle dérivé du modèle de Gepts pour le propofol. Il a été établi chez des adultes jeunes de classe ASA I ou II. La seule covariable prise en compte est le poids du patient.
Chez le sujet âgé ou fragile, ce modèle peut être utilisé à condition de réduire les concentrations-cibles et de réaliser une induction par titration.
Le modèle de Schnider
Il a été validé chez les patients allant de 18 à 81 ans et pesant de 44 à 123 kg. Il a été construit par une analyse pharmacocinétique de population avec comme covariables : l’âge, le sexe, la taille et la masse maigre. Le modèle de Schnider paraît intéressant chez le sujet âgé où le modèle de Marsh ne correspond pas. De plus, comme l’ont montré de nombreuses études, il permet en ciblant le site effet un meilleur contrôle de la vitesse d’induction.

Bibliothèque disponible dans la base Primea Orchestra : le modèle pharmacocinétique, ses variables de pondération et les valeurs des coefficients pour le patient actuel y sont consignés.
Le modèle de Gepts
Il s’agit du modèle du sufentanil. C’est un modèle tricompartimental, avec un large volume de distribution, une clairance élevée, une demi-vie terminale longue (15-20h) et une demi-vie contextuelle rapide (<60 min pour une perfusion >10h). Il a été validé dans une population allant de 14 à 68 ans et pesant entre 47 et 94 kg. Ce modèle est parfaitement linéaire et il n’a pas de covariables (âge, poids, masse maigre). Le délai d’obtention du pic d’action au site d’action est long (6minutes) et pour réduire la durée de ce pic, il est préférable de cibler le site d’action.
Le modèle de Minto
Le rémifentanil, de par ses propriétés pharmacocinétiques, est le plus adapté des morphiniques à l’administration en mode AIVOC. Au vu de sa rapidité d’action, il peut donc être utilisé en ciblant le site plasmatique et le site d’action. Il a été obtenu à partir d’une analyse pharmacocinétique chez des sujets âgés de 20 à 85 ans et de poids allant de 45 à 110 kg. Une influence de l’âge et de la masse maigre a été mise en évidence. C’est un modèle tricompartimental, avec un faible volume de distribution, une clairance métabolique très élevée, une demi-vie terminale de 50 à 60 minutes et une demi-vie contextuelle de 3-4 minutes, indépendante donc de la durée de perfusion.

Mais dans la pratique clinique, comment peut-on utiliser ces différents paramètres pharmacologiques pour s’adapter aux besoins du patient ?